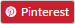
Regulatory writing is a branch of medical writing wherein the medical writer or the regulatory medical writer focuses on the documents that need to be submitted to the regulatory authorities at various stages of a clinical trial. At the start of the trial, regulatory documents like Protocol, Investigator Brochure (IB), and Informed Consent Form (ICF) need to be drafted. During the trial, there may be updates or amendments to any of these documents. Once the trial ends, Clinical Study Report (CSR) and common technical document (CTD) modules are required.
The medical writer need to be aware of all the regulations (of different countries) and also remain updated if there are any changes. These regulatory documents should transmit the required information accurately, transparently, and clearly to the target audience. These target audience varies according to the document type. For example- Regulatory authorities for eCTD Modules, Trial Investigators and ethics committee for Protocol, IB, CSR; and Patients for ICF.
The core skill set needed for regulatory writing remain fairly constant; understanding the target audience and regulation requirement, basic understanding of science, statistics, research; usage of grammar and writing/editorial skills, interpreting key messages, study design; project management and collaboration skills to work with reviewers, use of software, and having ethical standards.
As a specialist service provider in this area, we continue to provide content writing for various types of regulatory documents and have the necessary project management skills to cater to all the needs of our clients.
Kindly contact us for a confidential discussion of your project writing needs.